
I think this must be checkmate.
I’ve noticed that in the low-carb/cholesterol-denialism (yes, they are synonymous) community, much attention is now being given to Lp(a).
Not even attention – causal language:
“LDL-cholesterol isn’t causal, but Lp(a) is causal!”
If this is where they’re at, then truly the metaphorical long winter in denial retreat from Moscow has ended for them, and the rest of us can simply move on from the conversation safe in the knowledge that they have absolutely no understanding of what they’re talking about, either in terms of lipoproteins and cardiovascular disease (CVD) risk, or in relation to causality.

Figure illustrating desperate last stand of LDL-Denialism.
Let’s just take the latter first. Generally, the Bradford-Hill criteria is used as a conceptual framework to assess causality: strength of the association, consistency, specificity, temporality, biological gradient (dose-response), biological/mechanistic plausibility, coherence, reversibility, experimental evidence.
To be deemed ‘causal’, an exposure should meet each of these criteria, although some hold more weight than others, i.e., the presence of a strong dose-response where an exposure precedes the outcome (temporality), and where reversing the exposure reduces or eliminates incidence of disease, hold more weight than specificity (because biological systems are complex).
LDL-C is deemed causal because there isn’t a criteria you can come at it with in the evidence that isn’t satisfied by examining the mountain of evidence from:
- over 200 prospective cohort studies demonstrating a strong association and clear dose-response
- randomised controlled clinical intervention trials with a collective two-million participants
- 20-million person-years of follow-up during which over 150,000 CVD events have occurred
- Demonstrable reversibility in atherosclerosis from lowering LDL-C to <70mg/dL in primary prevention
- Mendelian randomisation studies on genetic predispositions to elevated or reduced lifelong exposure to LDL-C
I’m not going to rehash the argument in favour of LDL causality. I’ve gone through the history of the diet-heart hypothesis here, common fallacies and arguments deployed against it here, and a thorough step-wise run through the Bradford-Hill Criteria here. I’ve also gone through a comprehensive review of lipids and cardiovascular disease here, and another thorough rundown of the specific claims made in a debate with Dave Feldman here. Suffice it to say that I’m not sure there is much more I can contribute to the science communication of LDL-C causality in nutrition circles.
So is Lp(a) causal?
Nearly. For Lp(a), the epidemiology of the strength of the association and dose-response are evident, together with mechanistic understanding of the composition and functional properties of this lipoprotein subclass, and Mendelian randomisation studies inferring a causal role in atherosclerosis.
What is missing is an intervention trial directly testing the Lp(a) hypothesis, i.e., direct lowering of Lp(a). Lp(a) has been reduced in secondary analyses of PCSK9-inhibitor trials, which are glorious, but these trials were not directly targeting Lp(a) reduction specifically. For example, in the FOURIER trial, in which participants already had low LDL-C levels (~100mg/dL or less), the magnitude of risk reduction for coronary events was most significant in participants with high Lp(a) levels.
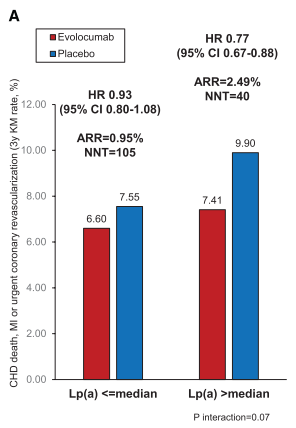
Figure from O’Donoghue et al. (Circulation. 2019;139(12):1483–92.) demonstrating reduction in coronary events in participants with higher Lp(a) levels following treatment with the PCSK9-inhibitor, evolocumab. A median reduction in Lp(a) of 26.9% was observed, with greater reductions in participants with higher baseline levels of Lp(a). A 23% relative risk reduction was demonstrated, with a significantly higher absolute risk reduction and greater than half the numbers needed to treat.
While these secondary analyses are suggestive of a benefit to direct reduction of Lp(a), there is currently no drug which specifically targets Lp(a) reduction. But these direct interventions are underway: the Lp(a)HORIZON trial is a Phase III clinical trial testing the effects of a new drug, TQJ230, on lowering Lp(a) in secondary prevention patients with Lp(a) >70mg/dL (not to be confused with the same level of LDL, they have different mass), with an estimated completion date of 2024. As a Sexy Drug Trial, it is aiming for ~8,000 participants. Nutrition science looks on in envy.
This would be the final piece in the causal puzzle for Lp(a): if direct lowering of Lp(a) reduces CVD events, then Lp(a) will join LDL-C as a causal risk factor for CVD. Of course, what the low-carb/cholesterol denialism community can’t seem to grasp is that more than one factor may be causal, while on the other hand being a causal risk factor doesn’t invalidate the importance of other systems biomarkers. Silly rabbits.
Anyway, I digress.
In the interim, while awaiting the results of large intervention studies, there are Mendelian randomisation studies. And this is where the argument that LDL-C is not causal, but Lp(a) is, wrecks on the rocks.
The first thing to note is that Lp(a) contains an LDL particle. Which makes the “Lp(a) is causal but LDL-C isn’t” claim almost absurd from the outset.
The ‘a’ in Lp(a) comes from the addition of the Apo(a) protein particle, in addition to ApoB, to the lipoprotein. A useful analogy I’ve seen for this is to imagine Apo(a) is like adding a Velcro patch to a normal LDL particle. It’s sticky, and makes it easy to get stuck in the artery wall.
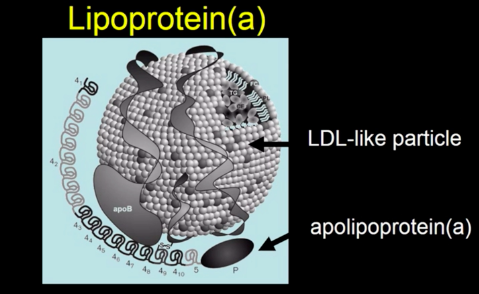
So, if Lp(a) contains an LDL particle with additional stickiness, where do they differ? In their mass concentrations (I’ll explain further). So how much would you have to lower Lp(a) to get a clinically meaningful reduction in heart disease risk?
Burgess et al. (JAMA Cardiology. 2018;3(7): 619-627) conducted a Mendelian randomization analysis involving >80,000 patients and>150,000 controls, where the genetic effect of lower Lp(a) levels was analysed and compared to the effects of lower LDL-C levels.
They demonstrated that the reduction in risk from lowering Lp(a) was proportional to the mass concentration, and that a reduction of 100mg/dL Lp(a) would be required to achieve a meaningful reduction in CHD risk. Now, here is where the rubber hits the road: the magnitude of this 100mg/dL reduction in Lp(a) was similar to the effect of lowering LDL-C by 38mg/dL – see the Figure from the study, below (LDL-C on the left, Lp(a) on the right).
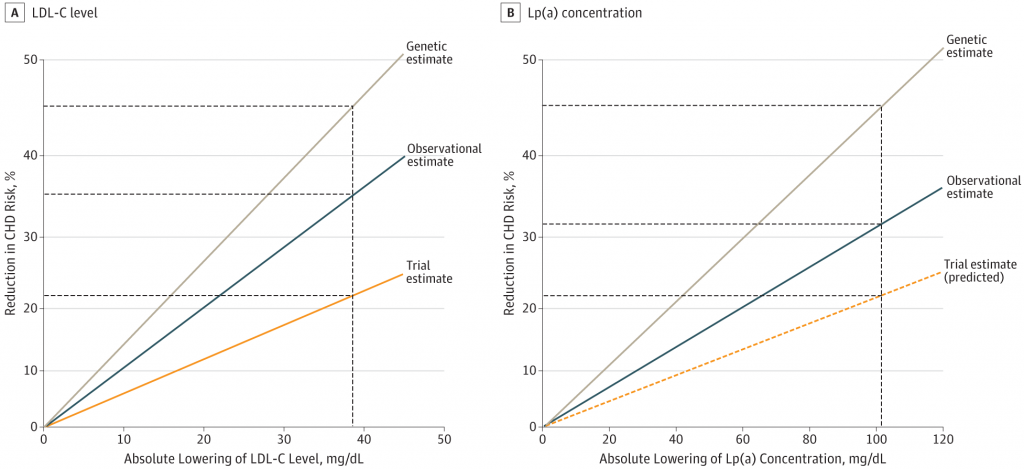
And why is this interesting? Because although their mass concentrations may differ – 100mg for Lp(a) and 38mg for LDL – the cholesterol content of these respective masses is essentially similar: 30-40%.
This in effect tells us what is known: that the transport of cholesterol into the artery, and deposition of cholesterol therein, is the initiating process of atherosclerosis.
Thus, although the mass concentrations between 100mg Lp(a) and 38mg LDL differ, the effect of reducing either by those respective masses is to reduce circulating cholesterol by roughly the same amount. And reduce CHD risk by roughly the same, as a result of lowering the causal exposure.
So for LDL-C denialists to argue that Lp(a) is causal is the most self-defeating cockology ever witnessed.
It is literally them arguing for the exact position – the lipoprotein properties, the mechanism of action, all of it – that they otherwise deny.
It’s amazing. Justice as only most poetic of bard’s could elucidate.
Either they actually don’t understand what the argument is about, don’t have a basic grasp of lipoproteins, don’t understand the mechanisms, or don’t understand causality in science.
Wait: it’s all of the above